Welcome to AutoREACTER
AutoREACTER v0.2.3
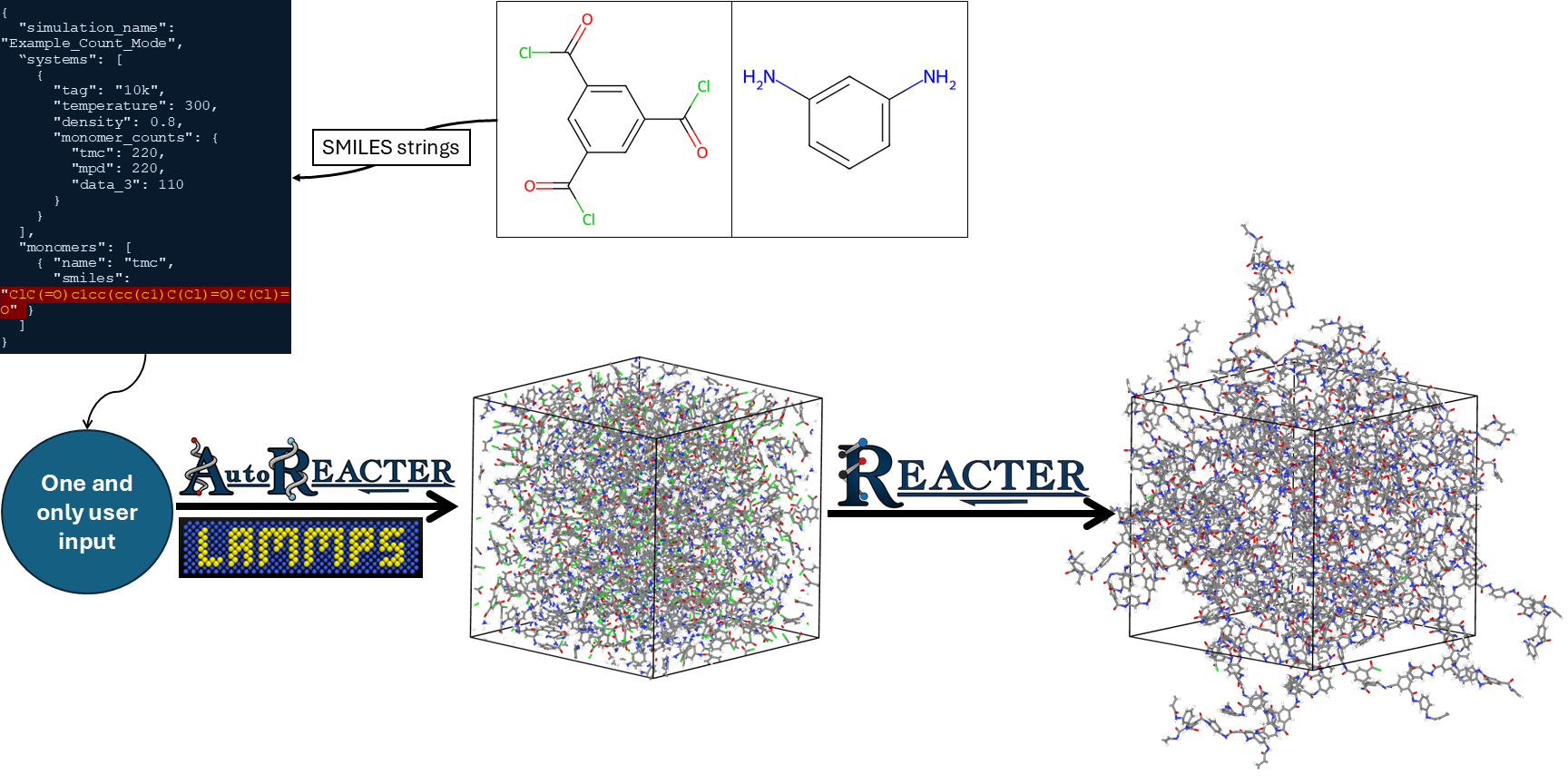
AutoREACTER is a Python-based toolkit for managing and automating reaction modeling in LAMMPS, for REACTER simulations. It provides a streamlined workflow for generating LAMMPS input files from simple chemical descriptions, eliminating the need for manual template preparation and atom typing.
Note: AutoREACTER is currently in v0.2.3. It is under active development, and APIs or functionality may change as we continue to expand the reaction library and force field support.
AutoREACTER is a Python-based toolkit designed to automate the setup and modeling of complex chemical reactions in LAMMPS. AutoREACTER bridges the gap between raw chemical structures and REACTER-ready LAMMPS input files for atomistic simulations.
For REACTER, you need to manually prepare molecule files, pre-reaction templates, post-reaction templates, and map files. Then, you must assign atom types to these files and write input scripts for LAMMPS.
AutoREACTER removes this manual process of defining reaction templates, preparing MAP files, and building topology files—using simplified input files as described in Input Configuration Documentation.
Advantages of AutoREACTER
Dual Workflows: Run automated pipelines via the CLI, or use interactive Jupyter Notebooks to visually validate intermediate steps, functional groups, and template mappings before writing a single LAMMPS file.
Automatic Reaction Detection: Molecules are first filtered based on functional group availability. Based on these functional groups, the necessary reaction templates for REACTER are automatically generated.
Automated Force Field Integration: Fully integrated with the LUNAR API to automatically assign partial charges for all reactant and product templates. Available force field options are selected through the input configuration workflow.
Smart File Generation: Automatically generates fully configured LAMMPS input scripts, including system densification, pre-equilibration, and multi-stage reaction files based on temperature, density, and monomer/molecule ratios or counts.
How AutoREACTER Works
AutoREACTER is built on a clean, modular, class-based architecture. When you submit your simulation setup, the toolkit processes it through five core stages:
Input Processing:
Reads yourinput.jsonfile and validates system simulations, temperatures, densities, monomer SMILES strings, stoichiometric ratios, and force field selections.Functional Group & Reaction Detection:
Analyzes monomers to identify available functional groups. These are cross-referenced with the internal reaction library to determine all possible reactive pathways. Non-reactive molecules are identified separately.Reaction Template Generation:
Uses RDKit and mapped SMARTS strings to generate pre- and post-reaction molecules and identify initiator atoms.
A 1:1 atom mapping is performed between pre- and post-reaction structures. Thewalker.pymodule traverses the molecular graph from initiator atoms to extract templates and edges. RDKit is also used to identify by-products throughout the reaction process.Force Field Assignment:
Communicates with the LUNAR API to assign force field parameters, atom types, and partial charges.
LUNAR repository: https://github.com/CMMRLab/LUNARLAMMPS File Generation:
Based on the provided inputs and generated chemical data, AutoREACTER writes the required LAMMPS input scripts.
Note: In the documentation and within AutoREACTER, molecules are often referred to as monomers, as this package is primarily designed for polymer systems.